
To use your whole genome file with Genetic Lifehacks, you’ll need to convert it into a format similar to raw data files from 23andMe or AncestryDNA. Once converted, your file will include all the SNPs found in those services, filling in all the genotypes included on Genetic Lifehacks.
Whole genome files come in multiple file formats, including:
- BAM/CRAM files
- VCF files
- FASTQ files
Options for whole genome file conversion:
Option 1) VCF conversion service
UGenome offers a VCF to TXT conversion service. Genetic Lifehacks members can use the coupon code GLH10 for $10 off the conversion (makes it $9). This works for TellMeGen and DNAComplete (Nebulus) VCF files.
Option 2) Genetic Lifehacks BAM/CRAM conversion service for members:
If you can’t use the WGSExtract software to do the conversion yourself: Genetic Lifehacks file conversion service
This is a service offered to members for $24.99.
Option 3) Convert the BAM or CRAM yourself for FREE
Directions below on how to use WGS Extract to convert your whole genome BAM or CRAM file.
Converting the BAM or CRAM file using WGS Extract:
This guide explains how to convert a whole genome BAM or CRAM file into a 23andMe-style raw data .txt file using a free tool called WGSExtract. The software can also convert some FASTQ files to a BAM/CRAM file and then to a raw data .txt file.
Request BAM for Sequencing:
If you used Sequencing.com, they no longer put the BAM file in your downloadable file list by default, but if you request it through their customer support, they will add your BAM file to your download list.
Hardware requirements:
To use WGS Extract, you will need a desktop or a fast laptop computer due to the file size and storage needed. File sizes for whole genome files are large (10-100GB), and you’ll need more than double that amount of storage space for the conversion. Windows, Mac, and Linux are supported.
Download the software:
WGS Extract is a free, open-source software that you can download from GitHub:
https://github.com/WGSExtract/WGSExtract.github.io
A detailed instruction manual is available on the GitHub page.
Read the manual:
Follow the installation instructions for your operating system. You’ll find the installation instructions at the end of the WGSExtract manual.
Yep, this is one of those times that you will need to read the manual. The software is a bit rough around the edges as far as the user interface and installation. The plus side is that the application is free, works well, and does exactly what is needed.
It takes a lot of free hard drive space to work with these large files. If you don’t have enough storage, you can use an external (USB) drive for the required storage space.
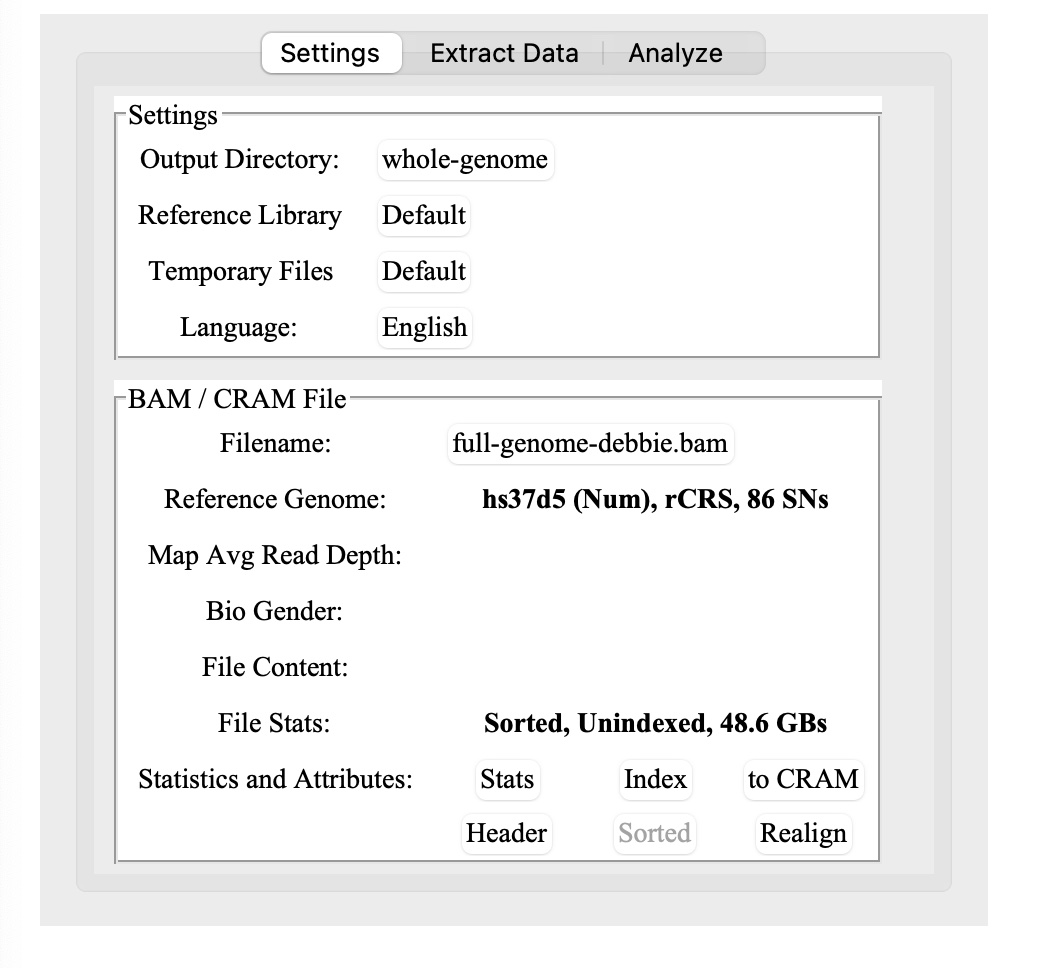
Once WGSE is installed and running:
1) Create a folder for the output files. It is recommended that the output folder not be the same folder as your data file is in. So, set up a new folder, and then select that folder for the Output Directory.
2.) Select your BAM file on your hard drive. (If you’re using Nebula or My Nucleus data, use the CRAM file)
3.) When you load in your BAM file, it may pop up an alert that it needs to be indexed. Sequencing.com or other .BAM files may already be indexed, so you could skip ahead to step 4.
Note: The reference library for a Dante Labs whole genome uses hs37d5.
Click on the Index button toward the bottom (next to where it says Statistics and Attributes:)
4.) Now, wait about 20-30 minutes or so to generate the BAM index file. You can do other things on your computer while waiting, but don’t close the application or the terminal window. For example, you can read through the manual again while you wait… :-)
5.) Key: Once you’ve indexed the BAM file, you will need to click on the Stats button before you can do anything else. Yes, it clearly says this in the manual, but I missed it and was confused for a bit.
6.) Next, click on the Extract Data tab at the top, and then on the Microarray RAW button.
Select “Combined file of ALL SNPs” for use on Genetic Lifehacks. There are many other options to come back later and play with if you are using the files for other genealogy sites.

7. Click the Generate button.
It will give you the expected wait time for the processing. Mine said 50 minutes, but it took about half that.
Once complete, your data files should be in the output folder that you set up for WGS Extract to use.
8. Back it up: Be sure to store your converted file safely and also back it up somewhere else.
You can now connect to the new file on the Member’s Dashboard.