Key takeaways:
~ Copper is necessary in small amounts, but too much copper can cause neurological and liver problems.
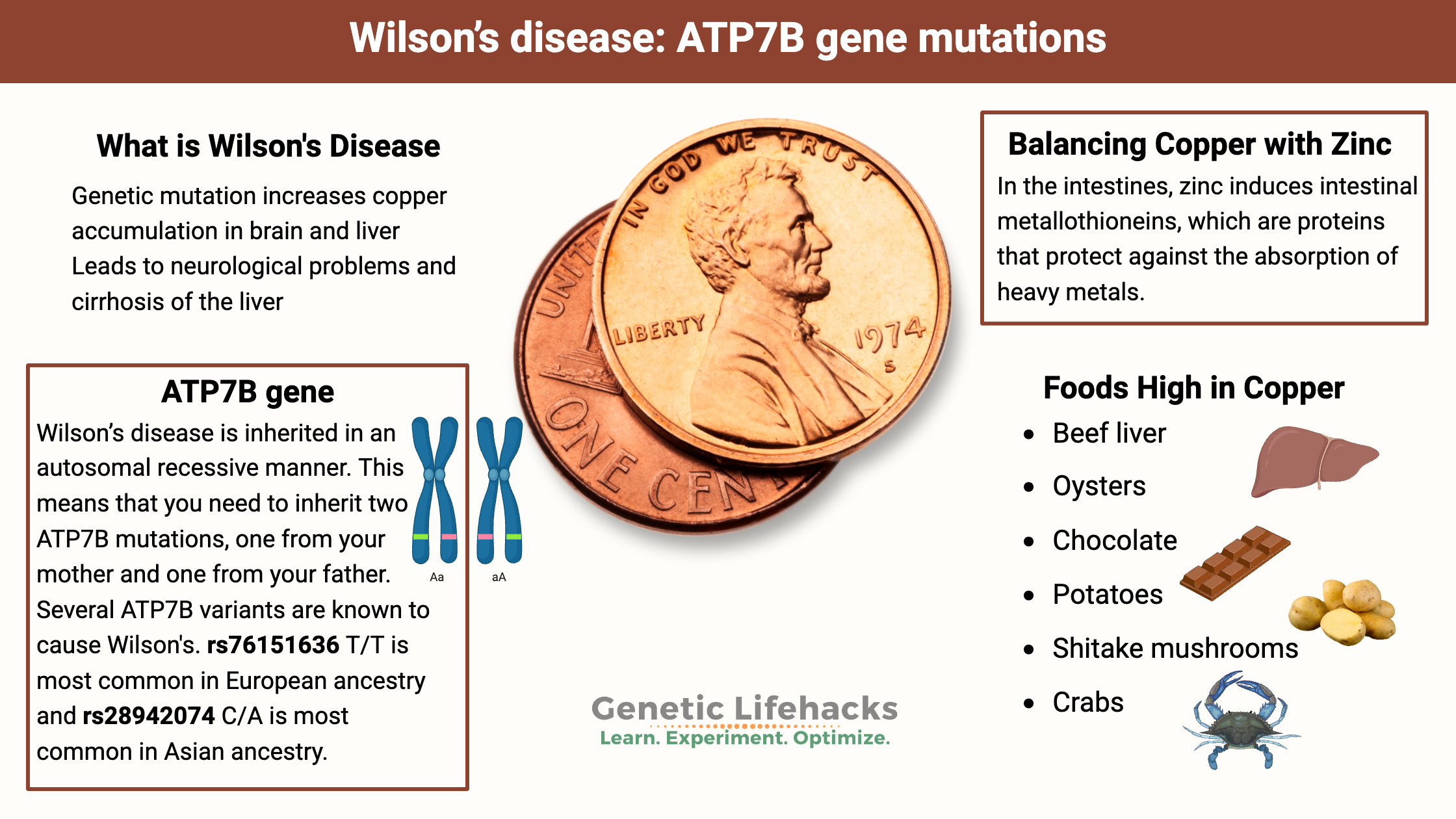
~ Mutations in the ATP7B gene can cause an excess of copper in the liver and brain. This is called Wilson’s disease.
~ About 1 in 90 people carry one copy of the mutation. While Wilson’s disease is usually only found in people with two copies of ATP7B mutations, people with one mutation may have more subtle changes to copper levels.
Wilson’s disease and copper accumulation:
Wilson’s disease is named after Samul Wilson, who published a paper in 1912 on the co-occurrence of neurological deficits and cirrhosis of the liver. In the 1940s, it was discovered that increased copper accumulation was the cause of the disease[ref]
Why do we need copper?
Copper is an essential trace mineral needed by the body in small amounts. The U.S. recommendation for adequate daily intake is 900 µg of copper for adults. The upper tolerable limit is set at 10 mg/day to avoid liver damage. According to the NIH, overt copper deficiency is uncommon and most people consume more than 1 mg of copper per day from food. However, it is estimated that up to 15% of people do not meet the recommended daily intake of 900 mcg of copper. People with celiac disease or who take high doses of zinc supplements are more likely to have decreased copper absorption[ref]
Copper is used in the mitochondria as part of the process of making ATP for cellular energy. Copper is also found in the enzyme superoxide dismutase (SOD), which acts as an antioxidant in cells.
Copper is primarily transported in the bloodstream by ceruloplasmin, an enzyme that contains copper and is thought to interact to facilitate iron transport. A smaller amount of copper circulates as free copper in serum.
Excess copper can cause problems in the body due to its oxidative capacity. Thus, copper levels are controlled both by limiting intestinal absorption and by chaperone proteins.[ref]
Symptoms of Wilson’s disease (WD):
Wilson’s disease is caused by genetic mutations in the ATP7B gene and can include neurological and/or liver symptoms. In Wilson’s disease, copper can accumulate in the liver tissue, resulting in lower levels of copper in ceruloplasmin. Excess copper in the liver can cause oxidative damage to cells, which can lead to fibrosis and cirrhosis.
However, not everyone with Wilson’s disease (WD) has the same symptoms.
Let’s take a closer look at the different ways copper affects the brain and liver.
Liver injury:
Jaundice and abnormal liver findings are characteristic of Wilson’s disease. Abdominal pain and vomiting may also be symptoms. An excess of free copper in the liver causes oxidative stress, which damages or kills cells. It can also cause lipid peroxidation in the liver. This can lead to mild to severe liver damage in some people with Wilson’s disease.[ref]
Ceruloplasmin and free copper:
Copper normally circulates bound to the protein ceruloplasmin. The Wilson’s disease mutations cause a decrease in ceruloplasmin, affecting the ratio of ceruloplasmin to free copper. Decreased serum ceruloplasmin levels are found in 95% of people with WD, and higher levels of unbound copper circulate.[ref]
Neurological symptoms:
Copper can also accumulate in the brain, causing neurological problems. A classic sign of Wilson’s disease is the wing-beating or flapping tremor, but other tremors may occur, as well as jerky dystonia. Involuntary grimacing or slow tongue movements may occur. Gait disturbances and fine motor problems may also occur.[ref]
Psychiatric symptoms can also occur in Wilson’s disease. For example, symptoms such as irritability, personality changes, disinhibition, anxiety, or depression are seen in WD. Cognitive impairment can be found in people with untreated Wilson’s disease.[ref]
Neurological symptoms of Wilson’s disease usually begin when people are in their 20s or 30s. However, it is possible to have late-onset Wilson’s disease (age 40 or older) or early-onset Wilson’s disease, which can occur in childhood.
Eye changes:
A Kayser-Fleischer ring, a copper or brown ring around the colored part of the eye, is seen in many patients with Wilson’s disease. This copper accumulation is reversible with copper chelation. However, Wilson’s disease is also associated with ocular motility problems and sunflower cataracts.[ref]
Ceruloplasmin levels <10mg/dl, along with Kayser-Fleischer rings, are often used to diagnose Wilson’s disease in the absence of genetic testing.
ATP7B gene: Copper transport in liver cells and ceruloplasmin
Mutations in the ATP7B gene cause Wilson’s disease in people who inherit two copies of the mutations. This is called autosomal recessive inheritance, meaning that people with WD inherited one ATP7B mutation from their mother and one mutation from their father.
ATP7B codes for a protein that regulates copper levels. It is found mainly in the liver, which plays an important role in tightly regulating the amount of free and bound copper in the body. The liver is responsible for excreting extra copper in the bile so that levels are controlled.
Copper is transported by ATP7B out of cells for excretion via the bile. Importantly, ATP7B is also required for the synthesis of functional ceruloplasmin, which transports bound copper in the serum.[ref] Ceruloplasmin is important for carrying copper in the bloodstream and for interactions with iron and transferrin. Both copper and iron can be oxidized when transported in their free state, and the body tightly controls how those metals interact with redox reactions.[ref]
Mutations in ATP7B can cause the protein to be made incorrectly or to have a shorter-than-normal half-life. As a result, copper is not properly transported within cells and is not effectively excreted from the body. Because of this, copper accumulates primarily in the liver, brain, and eyes due to the higher circulating free copper levels.[ref]
Dietary interactions:
Avoiding copper-rich foods is one dietary recommendation for WD patients, but it may be that diet is important in other ways as well.
Animal research and human case studies also show that a low-calorie diet may also help to prevent liver steatosis. In the liver, mitochondria are very susceptible to damage from copper overload, and mitochondria are also affected by metabolic changes, such as eating a lot of food. Animals fed a high-calorie diet had severe liver damage compared to the normal diet group, which had mild damage.[ref]
Age of onset and other genetic variants:
There is a wide variability in the symptoms of Wilson’s disease, which may be due to carrying other genetic variants along with two copies of the ATP7B mutations.
A 2011 study found that carriers of the MTHFR C677T or A1298C variants had an earlier age of onset of WD symptoms.[ref]
Another study found that males carrying the SOD2 rs4880 homozygous SNP were likely to have an earlier onset of symptoms.[ref]
Interactions with choline synthesis:
Choline is an essential nutrient that is used in the body in many different ways. One form of choline, called betaine or TMG, can donate a methyl group as part of the methylation cycle. Acetylcholine, an important neurotransmitter, is also synthesized from choline.
Animal studies led researchers to investigate how choline is disrupted in Wilson’s disease. A study comparing patients with Wilson’s disease to a healthy control group found that choline metabolites were significantly altered. The research showed that serum choline was elevated 4 times above normal in people with WD, and this was independent of any liver dysfunction. The conclusion was that the CDP-choline pathway is likely impaired in a way that is independent of copper overload.[ref]
Olfactory dysfunction in Wilson’s disease:
Olfactory dysfunction is also common in WD patients with neurological symptoms. One study using odor threshold discrimination tests along with MRI imaging found that there was neuronal loss in the olfactory region due to increased copper content.[ref] Another study found that the olfactory impairments were limited to specific odors.[ref]
What about having one copy of an ATP7B mutation?
It is estimated that 1-2% of the population carries one copy of an ATP7B mutation, called heterozygous carriers. People with one copy of an ATP7B mutation are not likely to have the extreme copper problems that people with Wilson’s disease have.
However, some studies do show slight differences due to the mutation. For example, studies show somewhat decreased serum ceruloplasmin concentrations in people who carry one copy of the mutation.[ref]
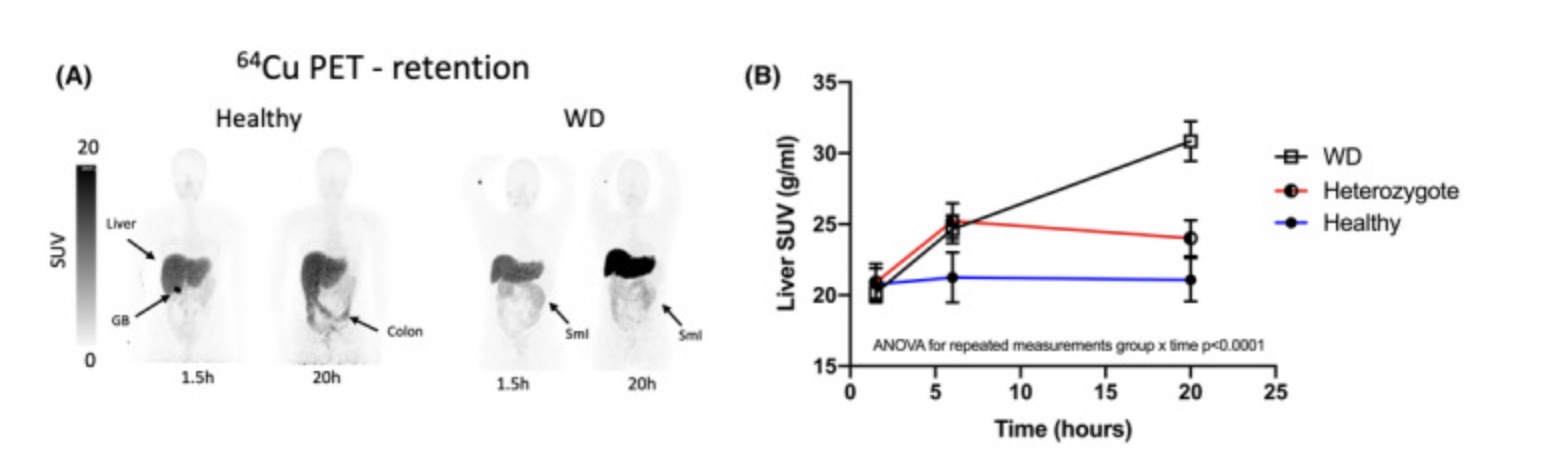
A study using PET/CT scans with a copper tracer looked at copper accumulation in normal adults (control group), adults with one copy of an ATP7B mutation, and adults diagnosed with Wilson’s disease (two ATP7B mutations). The results showed that after 20 hours, the Wilson’s disease patients retained significantly more of the copper tracer in their livers than the control group. People with one copy of the ATP7B mutation retained slightly more copper than the control group, but it was less than half the difference seen between the Wilson’s disease group and the control group.[ref]
Another imaging study found that people with one copy of an ATP7B mutation may accumulate free copper in the basal ganglia, which is the region of the brain involved in motor control and emotional behaviors.[ref]
Genotype report: ATP7B mutations
Wilson’s disease is inherited in an autosomal recessive manner. This means that you need to inherit two ATP7B mutations, one from your mother and one from your father. As I mentioned above, people who are carriers of one copy of a mutation may store a little more copper than normal and may have lower serum copper levels.
Cautions: 23andMe and AncestryDNA data is not guaranteed to be clinically accurate. For these and other rare mutations, please consider a clinical grade test to confirm before acting on the information. 23andMe and AncestryDNA data files do not cover all of the known ATP7B mutations, although they do cover the more common ones. Thus, you cannot rule out a mutation with the information here.
Access this content:
An active subscription is required to access this content.
Lifehacks:
Wilson’s disease is a serious condition that should be managed under a doctor’s care. There are medical treatments for copper chelation and tests your doctor can run to confirm what is going on.
The following information is presented for educational purposes for people who want to know more about copper levels.
Balancing copper with zinc:
Access this content:
An active subscription is required to access this content.
Related articles: