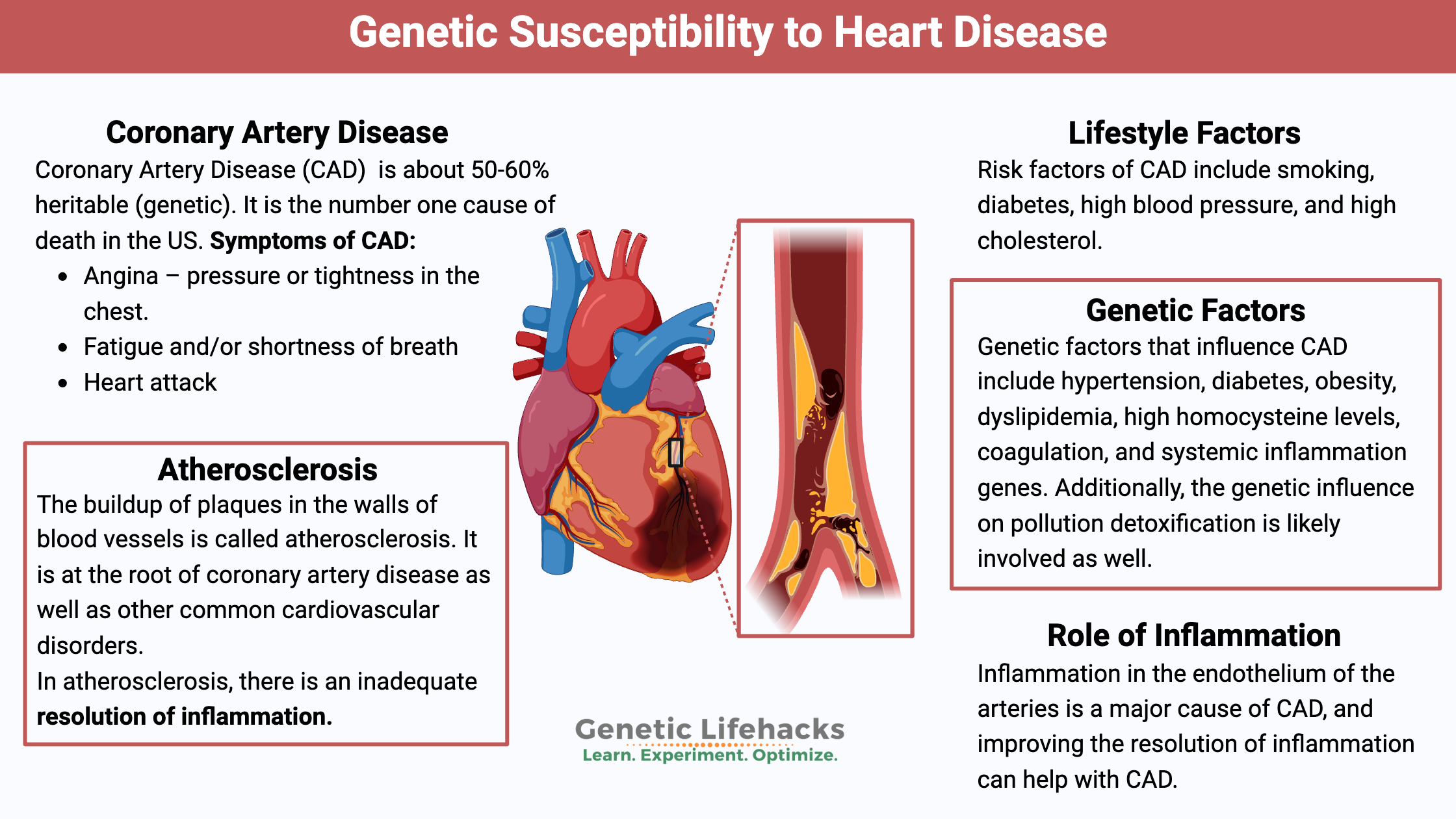
Key takeaways:
~ Heart disease is the number one cause of death, but there are a lot of different factors that can lead to heart disease.
~ Research shows that coronary artery disease is around 50% genetic, which means that both your lifestyle and your genes are involved.
~ Understanding the latest research on coronary artery disease and how your genes affect your risk can help you target your efforts for prevention.
~ Your genes can also give you a starting point to know which tests to get and what to talk with your doctor about.
Coronary Artery Disease (CAD): Genes, diet, lifestyle
Often called heart disease, coronary artery disease (CAD) is defined by doctors as developing when the major blood vessels to the heart become damaged. In general, atherosclerotic plaques in the arteries, along with inflammation, are blamed for CAD.[ref] As plaques build up, there is a narrowing of the arteries that bring blood flow into the heart. Coronary artery disease develops over the years, and it can take decades to become significant enough to notice.
Research shows us that coronary artery disease is about 50-60% heritable. It means that genetics is important, but so are lifestyle factors. For some of us, a ‘heart-healthy’ lifestyle is really important for preventing heart attacks and death.
Symptoms of CAD:
| Symptom | Description |
|---|---|
| Angina | Pressure or tightness in the chest |
| Fatigue/Shortness of Breath | General tiredness or difficulty breathing |
| Heart Attack | Sudden cardiac event |
If experiencing these symptoms, please be sure to talk with your doctor – or go to the ER for a heart attack. Your doctor can run tests such as an echocardiogram, stress test, or coronary artery calcium scan.
Prevalence:
Cardiovascular disease, a category that includes coronary artery disease, is the number one cause of death in the US and worldwide.
- About 18 million adults have coronary artery disease in the US, according to 2019 CDC data.[ref]
- Interestingly, cholesterol levels have declined from 1999 to 2016 across all the subgroups that are measured (gender, race, etc.).[ref]
- Myocardial infarction, or heart attack, can be mild or severe. About 50% of sudden deaths are due to myocardial infarction, with an average age of about 65 for men and 72 for women.[ref]
Lifestyle factors:
Always discussed with CAD are risk factors, including smoking, diabetes, high blood pressure, and high cholesterol. These are the big ones, but as you’ll see below, other factors can also increase CAD.
Genetic factors:
Researchers estimate that 50-60% of CAD risk is hereditary. Breaking this down, the research points to genetic factors that influence hypertension, diabetes, obesity, dyslipidemia, high homocysteine levels, coagulation, and systemic inflammation. Additionally, the genetic influence on pollution detoxification is likely involved as well.[ref]
What exactly is happening in coronary artery disease?
Health websites talk vaguely about CAD being caused by high LDL cholesterol from a bad diet, along with inflammation. For example, the Cleveland Clinic website explains, “If your cholesterol is too high, it builds up on the walls of your arteries. Over time, this buildup is known as atherosclerosis.”[ref]
You may be picturing fat from a cheeseburger sticking to the inside of your arteries and clogging them up. But… this isn’t what actually happens. A lot of what is written about high cholesterol and coronary artery disease is based on observations from over a hundred years ago, noting that the arteries had yellow streaks that looked like fat in them.
Enough of the generalities; let’s get to the heart of this issue (pun intended :-).
How do your arteries connect to heart disease?
Blood vessels are made up of several layers, and the innermost layer is the endothelium.
Between the endothelium and the outer wall of the blood vessel is where fatty, cholesterol-rich deposits can build up. It is called the sub-endothelium space.
Coronary artery disease is caused by the buildup of these cholesterol-rich plaques, called atherosclerotic plaque, in the arteries that supply the heart muscles with blood. When the heart muscle doesn’t get enough blood and oxygen, it cannot function well. It can result in angina (chest pain), fatigue from exertion, or even a heart attack.
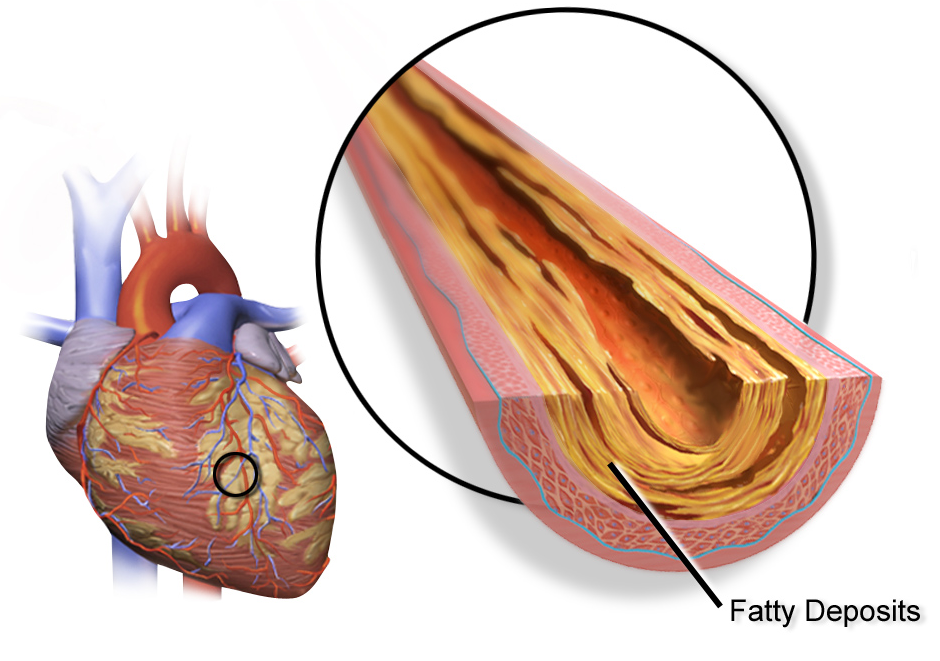
While it sometimes gets a bad rap, cholesterol is essential for your body to function. Cholesterol is a type of fat made in your body or consumed when eating animal-based foods.
- Cholesterol is an essential part of the membrane that surrounds the cell, and it is especially important in neurons in the brain. (Fun fact: 20% of your body’s cholesterol is in the brain.)[ref]
- Cholesterol is also essential for synthesizing hormones (testosterone, estrogen, cortisol, and vitamin D) and creating bile acids.
We all know that oil (fat) and water don’t mix. The same is true for cholesterol in the blood. Thus, cholesterol is packaged up with proteins and carried in the blood in molecules called lipoproteins.
Players in the CAD game:
Research shows us four “principal and interdependent processes” are involved in coronary artery disease:[ref]
| Process | Description/Key Points |
|---|---|
| Inflammation | Endothelial dysfunction, immune activation |
| Lipoprotein Handling | LDL, Lp(a), oxidized LDL, ApoE, PCSK9 |
| Endothelial Integrity | Role of nitric oxide, stressors, resolution |
| Thrombosis | Plaque rupture, clot formation |
These four processes are individually important and also intertwined.
Starting with inflammation in the endothelium:
The endothelium is the layer of cells that line blood vessels (and lymphatic vessels). It is a single layer of cells, and doctors used to think of the endothelium as just a lining, like a cellophane wrapper inside the blood vessels.
However, research over the last couple of decades shows that the endothelium has many vital functions, including secreting hormones, reacting to pathogens, controlling platelet activation, and regulating vascular tone.[ref]
In other words, it is nothing like a cellophane wrapper – instead, the endothelium runs the show. When the endothelium is healthy, such as in younger people, the cells are tightly joined, forming a barrier that LDL-cholesterol cannot cross.
One research paper explains: “endothelial cells (ECs) serve as multifunctional biosensors that coordinate vascular responses to environmental stress of which hypoxia, oxidative stress, acidosis, and inflammation are especially prominent in myocardial disease and cancer “[ref]
Let’s dig into these endothelial stressors:
- Acidosis is when your blood pH is off balance – too much acid or too little base (bicarbonate). It can occur when you have too much glucose and not enough insulin, especially in people with diabetes. Additionally, acidosis can be due to respiratory disease, tumors, or inflammation.[ref]
- Hypoxia is a lack of oxygen in cells. It can occur when blood vessels are narrowed, and not enough oxygen is carried through to meet demand.
Research shows that hypoxia, along with acidosis, triggers an inflammatory response in endothelial cells, including HIF-1a and HMGB1.[ref]
HIF-1a (hypoxia-inducible factor 1a) is a regulator of hundreds of other genes that respond to low oxygen. Activating HIF-1a in the short term causes some protective pathways to be initiated. But long-term activation of HIF-1a leads to cardiovascular dysfunction. HMGB1 is another protein that activates other genes – specifically, it amplifies an inflammatory response.[ref]
Related article: HIF-1A genetic variants
Research shows that HMGB1 (activated by hypoxia and acidosis) causes LDL-cholesterol transcytosis. When the endothelium is healthy, the LDL cholesterol can’t pass between the cells to get to the area between the endothelium and the arterial wall. The only way that LDL-c can get into that space with a healthy endothelium is via transcytosis – being taken into the endothelial cell and then deposited back out the other side. Thus, the HMGB1 alarmin causes the initiation of cholesterol moving into the area between the endothelium and the arterial wall without the endothelial junctions being loosened.[ref]
Related article: HMBG1 genetic variants
Other causes of endothelial inflammation include exposure to toxicants and pathogens. Your blood circulates everything you’re exposed to, and thus, the endothelial cells are exposed. Smoking is an easy example here: Smoking cigarettes dumps toxicants such as benzene, formaldehyde, and toluene into the lungs, which then transfer the harmful substances into the bloodstream. Similarly, viruses, bacteria, or pathogen parts can circulate in the bloodstream and interact with the endothelium.[ref] (More on this below)
Resolving Inflammation:
Doctors and researchers used to think that an inflammatory state resolved back to normal when the inflammatory markers just passively went away. It is why so many anti-inflammatory drugs, such as COX2 inhibitors, target the initial inflammatory cytokines.
But it turns out inflammation resolution is an active process, not a passive one. The resolution of inflammation involves anti-inflammatory mediators called lipoxins, resolvins, maresins, and protectins.[ref] The inflammation resolution molecules are derived from DHA and EPA (omega-3 fatty acids found in fish oil).[ref]
Related article: The Resolution of Inflammation and SPMs
Atherosclerosis: Plaque in your arteries
The buildup of plaques in the walls of blood vessels is called atherosclerosis. It is at the root of coronary artery disease as well as other common cardiovascular disorders.
Classically, the cholesterol hypothesis of atherosclerosis explains that elevated LDL is directly associated with atherosclerosis. The literature on this states that infiltration of apoB particles that contain LDL-cholesterol is the initial cause of atherosclerosis. This change starts with arterial injury, which causes the endothelium to be infiltrated by monocytes and apoB particles into the artery wall.[ref]
For atherosclerosis to occur, you need increased cellular adhesion molecules, which are found on endothelial cells that are activated by inflammatory cytokines. Increased cellular adhesion allows immune system molecules, such as macrophages, to adhere to endothelial tissue.[ref]
Inflammation is, of course, a natural and completely necessary response to injury or toxic insult. The key is that there needs to be a balance between the inflammatory process and the resolution of inflammation. In atherosclerosis, there is an inadequate resolution of inflammation in a timely fashion.[ref]
Macrophages, foam cells, and plaques:
The imbalance of too much inflammation or too little resolution is the heart of coronary artery disease and atherosclerosis. The body’s immune response is key, and several immune system cell types are important here. (Interestingly, mice bred to have immunodeficiency never get atherosclerosis.[ref])
Macrophages are immune cells that colonize tissues in the body. Being ’tissue-resident immune cells’ means that they aren’t roaming around your whole body, but rather specific forms of macrophages are on patrol in different tissues (lung, brain, liver, etc.).
Macrophages engulf and digest cells that don’t seem to be healthy. It includes cancer cells, foreign substances, viruses, bacteria, and cellular debris. In addition to defending our body against pathogens, macrophages also increase inflammation, stimulate other parts of the immune system, and encourage tissue repair.
Foam cells form when macrophages internalize ApoB-containing lipoproteins. The macrophage infiltration then increases oxidative stress and cytokine production, resulting in more endothelial cell activation, cellular adhesion, and recruitment of more macrophages.[ref]
It results in a build-up of macrophages and entrapment in the atherosclerotic plaque.
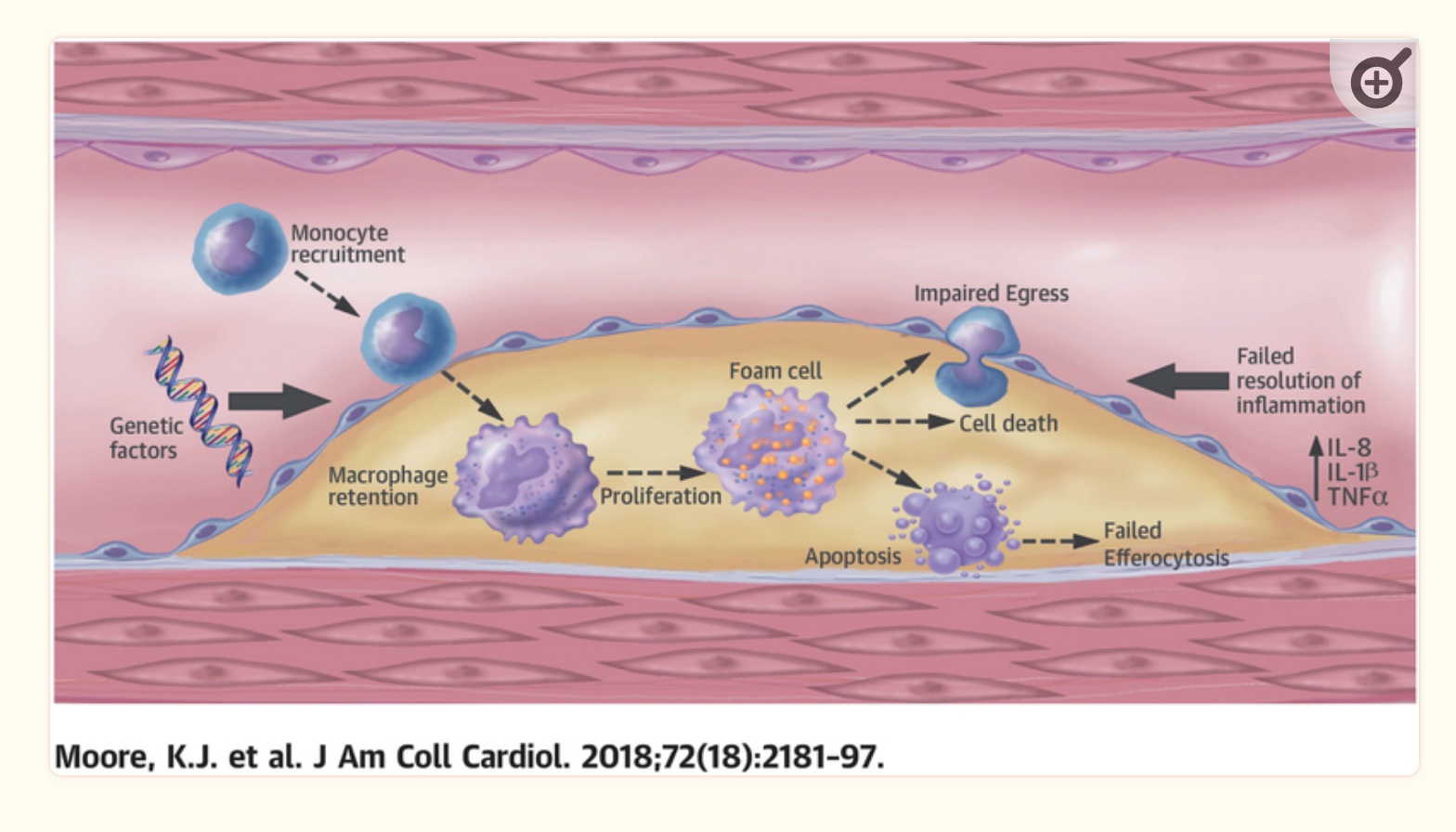
Dual role:
While macrophage infiltration is inflammatory, macrophages also play a role in resolving inflammation. Using DHA as a substrate, macrophages can biosynthesize maresin, a pro-resolvin, from DHA.[ref]
Macrophages can be divided into groups:[ref]
- M1 macrophages are classically activated (e.g., by pathogens) and sustain inflammation.
- M2 macrophages are alternatively activated and resolve inflammation.
- Mox macrophages are a proatherogenic subset responding to oxidized phospholipids.
Plaques to Clots to Heart Attacks:
Excessive inflammation due to entrapped macrophages and sustaining inflammation can lead to the formation of atherosclerotic plaques. The atherosclerotic plaque can eventually rupture, causing essentially a little tear in the blood vessel, which then is repaired by the body’s clotting cascade. The clot, if it is large enough, can block the blood vessel and restrict blood flow — not good when you are talking about blood flow to the heart muscle.
While macrophages and inflammation are at the heart of atherosclerosis, macrophages are also the solution. Animal studies dating back decades show that changing diet from a high-fat, high-cholesterol diet to one that is low-fat, low-cholesterol can reduce the thickness of the coronary artery plaques. Human studies show that lowering cholesterol helps, but perhaps not to the same degree as seen in animal studies.[ref] More on this in a bit…
The role of LDL cholesterol in cardiovascular disease:
Cholesterol hypothesis of heart disease: What does LDL cholesterol have to do with endothelial inflammation? And why does it end up in the wall of the blood vessel?
Essentially, the hypothesis is that either:
- Too much LDL causes it to end up in the arterial wall -or-
- Excessive oxidation of LDL causes the inflammatory cycle
Oxidized LDL-c and also LDL-cholesterol bound to glycosaminoglycans is recognized by macrophages as ‘foreign’, triggering an immune response.[ref]
As I mentioned above, research from 2019 on inflammation in the endothelial cells shows that an increase in HMGB1, an alarmin that increases other inflammatory cytokines, causes LDL cholesterol to be taken up by endothelial cells via endocytosis and then moved to the space between the endothelium and the arterial wall.[ref]
While these may seem contradictory – is it high LDL-c? or oxidized LDL-c? or endothelial inflammation? – I think all this can be true. Endothelial inflammation moves the LDL-c, and higher levels of LDL-c would then result in more of it being moved. LDL-c is oxidized by exposure to higher oxidative stress in the area of inflammation. To compound the issue, oxidized LDL-c is recognized by macrophages as ‘foreign’ and initiates an inflammatory response, driving the process of more inflammation, more cholesterol deposited, and more macrophages activated.
Thus, lowering really high LDL-c levels means that there is less of it to move across the endothelial cells by transcytosis. Lower levels of LDL-c mean that there is less of it available to be oxidized. Additionally, antioxidant therapy such as vitamin E or Vitamin C may help to reduce oxidized LDL-c. Finally, reducing inflammation along with increasing the resolution of inflammation (e.g., aspirin therapy) also helps.
Cholesterol and lipoproteins:
Cholesterol is a general term, and there are different lipoprotein particles, different sizes of LDL particles, etc., that all interact here.
- The different lipoprotein particles play different roles in cholesterol transport. For example, lipoprotein(a) is strongly linked to an increased risk of heart attacks. Lp(a) consists of LDL-c along with apo(a) and apoB100 particles. The apo(a) in Lp(a) blocks the resolution of clots and increases pro-inflammatory events in the cell wall.[ref]
- APOA5 gene variants link high triglycerides and higher ox-LDL levels to increased risk of CAD.[ref]
- Receptors for oxidized LDL are important, such as LOX-1[ref]
- Other receptors, such as the apoE2 receptor, are essential in reducing clotting and increasing blood vessel dilation.
Let me go into a little more detail on a couple of important regulators of cholesterol levels so that the genetics part (below) makes more sense:
1) APOE and receptors:
Apolipoprotein E (ApoE) is well studied in relation to Alzheimer’s risk. The APOE E4 variant significantly increases the risk of Alzheimer’s disease. ApoE is a lipoprotein that binds to and transports cholesterol. It is present in several different classes of lipoproteins.
A receptor for apoE, called the apolipoprotein E receptor-2, was identified in a genome-wide association study as being important in developing premature CAD. This prompted research into why an apoE receptor would influence the development of coronary artery disease. The receptor is found abundantly in the brain (important in memory formation) and testes (important in selenium uptake and fertility). At lower levels, the receptor was also found in platelets, endothelial cells (lining the blood vessels), and monocytes/macrophages. Animal research shows that when apoE binds to the apoE2 receptor, it stimulates nitric oxide (a vasodilator) and inhibits cellular adhesion. In other words, it makes the blood vessels dilate and prevents stickiness, such as found in a clot. The receptor also modulates macrophage activation and influences the formation of atherosclerosis.[ref]
Related article: APOE type (only read if you want to know your Alzheimer’s risk)
PCSK9 and atherosclerosis:
One way cells regulate the amount of LDL cholesterol is by taking in LDL-c through an LDL receptor. PCSK9 is a protein that causes fewer LDL receptors to be available on the surface of cells. Higher PCSK9 levels lead to higher LDL-c levels. Genetics plays a significant role in PCSK9 levels, with some variants leading to high LDL-c and other variants causing lifelong lower LDL-c.
While most PCSK9 is produced in the liver and kidneys, the endothelial cells lining blood vessels also produce PCSK9. Additionally, macrophages can produce PCSK9. It makes PCSK9 (along with LDL-c) an important player in the formation of atherosclerotic plaques. Animal studies show that PCSK9 has pro-inflammatory behavior in macrophages, increasing the formation of foam cells and plaque. When the PCSK9 gene is deleted in mice, it is protective even in animals that normally would have atherosclerosis.[ref]
Related article: PCSK9 and cholesterol
Calcium and hardening of the arteries:
In addition to inflammation and cholesterol infiltration, calcium also plays a role in heart disease.
In atherosclerosis, the thickening of the artery wall is a problem only when the blood vessel can no longer expand. As long as the artery can expand and compensate for the fatty deposits, blood flow is not reduced.
The problem of reduced blood flow comes when the blood vessel flexibility is reduced. This inflexibility is where calcium comes in. Calcification of the vascular smooth muscle cells reduces flexibility, but it also stabilizes the atherosclerotic plaque, hardening it and making it less likely to rupture and cause a heart attack.
You may have heard of a coronary calcium score, which measures calcium-containing plaques in your arteries.
Researchers have found two seemingly contradictory findings: highly calcified lesions are less likely to rupture, but a high calcium score is linked to a greater risk of future coronary events. Both seem to be true. Higher levels of calcification can stabilize large plaques, but calcium is also linked to fragmented or microcalcifications, which also cause coronary events.[ref]
Genetic variants linked to higher serum calcium levels are also linked to an increased risk for CAD.[ref]
Triggers of endothelial injury:
I want to circle back to the inflammation of the endothelium. What starts this whole process off?
There are a lot of causes of endothelial inflammation: bacteria, viruses, toxic substances, high blood glucose levels, hypoxia, autoimmune responses, shear stress from high blood pressure, and hyperlipidemia (high cholesterol, oxidized cholesterol, or really high triglycerides).[ref]
Keep in mind that endothelial injury isn’t a one-time event. You don’t end up with CAD or atherosclerosis from just a single hit. Your cells are resilient, so we can see inflammation occur and then resolve. It is the repeated and sometimes constant barrage of insults that causes long-term damage.
| Trigger/Factor | Mechanism/Effect |
|---|---|
| Smoking | Toxins damage endothelium |
| Microplastics | Increase inflammation, found in plaques |
| High Blood Glucose | Alters pH, damages endothelium |
| Pathogens | Bacteria/viruses disrupt endothelial function |
| Autoimmune Disease | Chronic endothelial injury |
| Toxicants | Chemicals, heavy metals |
Let’s dig into a couple of these triggers:
Microplastics:
New studies show that small, microscopic particles of plastic are being absorbed into the bloodstream and interacting with the endothelium. A 2024 study looked at 17 artery samples from surgery and found microplastics in all of the atherotic plaque samples.[ref] Another 2024 study found that acute coronary syndrome patients had elevated microplastic concentrations compared to a control group. There was a significant relationship between higher levels of microplastics and increased inflammation (IL-6, B cells, and natural killer cells).[ref] This makes sense as an environmental cause of endothelial inflammation.
Related article: Microplastics Research Roundup
Autoimmune disease:
Systemic sclerosis and Raynaud’s disease are both linked to endothelial injury that can lead to long-term changes in the blood vessels.[ref]
- Related article: Raynaud’s syndrome
High blood glucose:
Diabetes and cardiovascular disease go together for a reason. Severely high blood glucose levels alter pH, causing damage response and inflammation. But for most people with higher blood glucose levels, the damage to endothelial cells comes from the long-term accelerated degradation of nitric oxide (NO). NO is important in protecting cells from inflammation as well as relaxing the blood vessels.[ref]
Related articles: Type 2 Diabetes and Blood glucose levels
Toxicants and heavy metals:
We are exposed to all kinds of chemical toxicants daily, and our body does a good job of dealing with these insults by breaking them down and excreting them. But chronic exposure can also damage endothelial cells. Cigarette smoking is an excellent example, and several substances in cigarette smoke are well known for their ability to injure endothelial cells. Heavy metal exposure, such as mercury or lead, can also damage the endothelium, and certain medications (e.g., chemotherapy drugs) also cause damage. For example, lead exposure is directly tied to endothelial injury and an inhibition of endothelial repair.[ref][ref][ref]
Related article: Getting the Lead Out and Detoxifying Mercury
Pathogens:
Viruses and bacteria can invade endothelial cells. For example, the dengue virus can disrupt the barrier that protects the endothelial cells (called the glycocalyx) and cause havoc with the endothelium. Severe dengue fever causes ‘vascular leakage’.[ref] Ebola and other hemorrhagic fever viruses also disrupt endothelial function, but these are short-term events (often ending abruptly in death) and are not the cause of your atherosclerosis. Periodontal disease, though, is linked to endothelial dysfunction and cardiovascular disease. Chronic, low-grade inflammation due to gingivitis downregulates nitric oxide, eventually leading to coronary endothelial dysfunction.[ref][ref] The bacteria that cause periodontal disease, P. gingivalis, has also been found in atherosclerotic plaque.[ref]
Related article: Gingivitis and genetic susceptibility
Spike protein (from SARS-CoV-2) causes endothelial dysfunction:
The spike protein on SARS-CoV2 increases endothelial cell ROS, triggering autophagy and apoptosis. Recent research shows that the spike protein alone can also induce cell death in vascular endothelial cells by triggering an inflammatory response.[ref] One study found that the spike protein was more likely to cause endothelial injury “under conditions of exposure to androgen dihydrotestosterone (DHT) and tumor necrosis factor-a (TNF-α)”.[ref] Men, especially younger men, have higher overall androgen levels, but as men age, DHT tends to increase compared to testosterone.[ref]
While there are still a lot of unknowns with the SARS-CoV-2 spike protein, thrombocytopenia and thrombosis are rare side effects, especially in the adenoviral vector vaccines.[ref]
Making Connections: Genetic susceptibility in context
Below, you’ll see your genetic variants that are directly tied to increased susceptibility to CAD. However, I wanted to also make clear that heart disease isn’t a standalone condition. Your genetic susceptibility to inflammation, not resolving inflammation, detoxification, and immune response, all can play a role.
| Factor | Connection |
|---|---|
| Resolution of Inflammation | Not having enough DHA/EPA for SPM synthesis, and genetic variants that impact conversion to SPMs |
| HIF-1A | Variants in HIF1A gene alter response to hypoxia, which is integral to CAD |
| HMBG1 | HMBG1 genetic variants increase the baseline inflammatory response to acidosis and hypoxia |
| APOE type | The APOE type affects both Alzheimer’s and heart disease risk in conjunction with diet |
| Raynaud’s syndrome | Genetic susceptibility plays a role in Raynaud’s, which can then lead to endothelial injury |
| Gingivitis and genetic susceptibility | SNPs affect whether you are likely to host P. gingivalis, which promotes the formation of atherosclerotic plaque |
| Blood glucose levels | Genetic variants influence blood glucose levels, even in the absence of diabetes, which can damage the endothelium and lead to atherosclerosis |
| Detoxifying Mercury | SNPs affect how well you detoxify mercury, and mercury directly damages the endothelium. |
| Detoxifying Lead | Variants that decrease your ability to detoxify lead could allow for endothelial damage at lower lead exposure levels. |
Coronary Artery Disease Genotype Report:
How can you use this information?
The genetic variants below will explain some of your relative risk for coronary artery disease. If you have a higher than normal risk of CAD, lifestyle factors (healthy diet, exercise, sleep) will be important for you in keeping your heart healthy.
Lifehacks for preventing heart disease:
Doctors tell you that heart health comes from eating a good diet, exercising, being at the right weight, and not smoking.
All good things to do… But what does it mean to eat a good diet? How much do an extra 20 pounds matter? Are there other changes or supplements that can give you the most bang for your buck?
Below is information on recent research studies on CAD and atherosclerosis. As always, talk with your doctor for specific medical advice.
Consider joining today to see the rest of this article.
Related Articles and Genes:
References:
Abraham, David, and Oliver Distler. “How Does Endothelial Cell Injury Start? The Role of Endothelin in Systemic Sclerosis.” Arthritis Research & Therapy, vol. 9, no. Suppl 2, 2007, p. S2. www.ncbi.nlm.nih.gov, https://doi.org/10.1186/ar2186.
—. “How Does Endothelial Cell Injury Start? The Role of Endothelin in Systemic Sclerosis.” Arthritis Research & Therapy, vol. 9, no. Suppl 2, 2007, p. S2. www.ncbi.nlm.nih.gov, https://doi.org/10.1186/ar2186.
Anderson, Jeffrey L., et al. “Genetic Variation at the 9p21 Locus Predicts Angiographic Coronary Artery Disease Prevalence but Not Extent and Has Clinical Utility.” American Heart Journal, vol. 156, no. 6, Dec. 2008, pp. 1155-1162.e2. PubMed, https://doi.org/10.1016/j.ahj.2008.07.006.
Barale, Cristina, et al. “PCSK9 Biology and Its Role in Atherothrombosis.” International Journal of Molecular Sciences, vol. 22, no. 11, June 2021. www.ncbi.nlm.nih.gov, https://doi.org/10.3390/ijms22115880.
Barrett, Tessa J. “Macrophages in Atherosclerosis Regression.” Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 40, no. 1, Jan. 2020, p. 20. www.ncbi.nlm.nih.gov, https://doi.org/10.1161/ATVBAHA.119.312802.
Benjamin, Emelia J., et al. “Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association.” Circulation, vol. 139, no. 10, Mar. 2019, pp. e56–528. ahajournals.org (Atypon), https://doi.org/10.1161/CIR.0000000000000659.
Bilotta, Clio, et al. “COVID-19 Vaccine-Related Thrombosis: A Systematic Review and Exploratory Analysis.” Frontiers in Immunology, vol. 12, 2021. www.ncbi.nlm.nih.gov, https://doi.org/10.3389/fimmu.2021.729251.
“Coronary Artery Disease – Symptoms and Causes.” Mayo Clinic, https://www.mayoclinic.org/diseases-conditions/coronary-artery-disease/symptoms-causes/syc-20350613. Accessed 19 Jan. 2022.
Dai, Xuming, et al. “Genetics of Coronary Artery Disease and Myocardial Infarction.” World Journal of Cardiology, vol. 8, no. 1, Jan. 2016, p. 1. www.ncbi.nlm.nih.gov, https://doi.org/10.4330/wjc.v8.i1.1.
Dong, Lixue, et al. “Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells.” International Journal of Molecular Sciences, vol. 18, no. 2, Jan. 2017, p. E278. PubMed, https://doi.org/10.3390/ijms18020278.
Duvall, Melody G., and Bruce D. Levy. “DHA- and EPA-Derived Resolvins, Protectins, and Maresins in Airway Inflammation.” European Journal of Pharmacology, vol. 785, Aug. 2016, p. 144. www.ncbi.nlm.nih.gov, https://doi.org/10.1016/j.ejphar.2015.11.001.
Enas, Enas A., et al. “Lipoprotein(a): An Underrecognized Genetic Risk Factor for Malignant Coronary Artery Disease in Young Indians.” Indian Heart Journal, vol. 71, no. 3, June 2019, p. 184. www.ncbi.nlm.nih.gov, https://doi.org/10.1016/j.ihj.2019.04.007.
Guionaud, Silvia. “The Far Side of Vascular Injury: Nonconventional Vasoconstrictors, DNA-Targeting Agents, and Agents Toxic to Vascular Smooth Muscle.” Toxicologic Pathology, vol. 43, no. 7, Oct. 2015, pp. 945–58. SAGE Journals, https://doi.org/10.1177/0192623315601905.
Herrero-Fernandez, Beatriz, et al. “Immunobiology of Atherosclerosis: A Complex Net of Interactions.” International Journal of Molecular Sciences, vol. 20, no. 21, Nov. 2019. www.ncbi.nlm.nih.gov, https://doi.org/10.3390/ijms20215293.
“High Cholesterol Diseases: Conditions & Outcome.” Cleveland Clinic, https://my.clevelandclinic.org/health/articles/11918-cholesterol-high-cholesterol-diseases. Accessed 19 Jan. 2022.
Hussain, Mehak, et al. “P. Gingivalis in Periodontal Disease and Atherosclerosis – Scenes of Action for Antimicrobial Peptides and Complement.” Frontiers in Immunology, vol. 6, 2015. www.ncbi.nlm.nih.gov, https://doi.org/10.3389/fimmu.2015.00045.
Interactive Atlas of Heart Disease and Stroke Tables. https://nccd.cdc.gov/DHDSPAtlas/Reports.aspx. Accessed 19 Jan. 2022.
Jin, Uram, et al. “Cholesterol Metabolism in the Brain and Its Association with Parkinson’s Disease.” Experimental Neurobiology, vol. 28, no. 5, Oct. 2019, p. 554. www.ncbi.nlm.nih.gov, https://doi.org/10.5607/en.2019.28.5.554.
Jinnouchi, Hiroyuki, et al. “Calcium Deposition within Coronary Atherosclerotic Lesion: Implications for Plaque Stability.” Atherosclerosis, vol. 306, Aug. 2020, pp. 85–95. PubMed, https://doi.org/10.1016/j.atherosclerosis.2020.05.017.
Kasikara, Canan, et al. “The Role of Non-Resolving Inflammation in Atherosclerosis.” The Journal of Clinical Investigation, vol. 128, no. 7, July 2018, p. 2713. www.ncbi.nlm.nih.gov, https://doi.org/10.1172/JCI97950.
Kim, Minjoo, et al. “A Promoter Variant of the APOA5 Gene Increases Atherogenic LDL Levels and Arterial Stiffness in Hypertriglyceridemic Patients.” PLoS ONE, vol. 12, no. 12, 2017. www.ncbi.nlm.nih.gov, https://doi.org/10.1371/journal.pone.0186693.
Kohli, Payal, and Bruce D. Levy. “Resolvins and Protectins: Mediating Solutions to Inflammation.” British Journal of Pharmacology, vol. 158, no. 4, Oct. 2009, p. 960. www.ncbi.nlm.nih.gov, https://doi.org/10.1111/j.1476-5381.2009.00290.x.
Kumar, Nitin, et al. “SARS-CoV-2 Spike Protein S1-Mediated Endothelial Injury and Pro-Inflammatory State Is Amplified by Dihydrotestosterone and Prevented by Mineralocorticoid Antagonism.” Viruses, vol. 13, no. 11, Nov. 2021, p. 2209. PubMed, https://doi.org/10.3390/v13112209.
Larsson, Susanna C., et al. “Association of Genetic Variants Related to Serum Calcium Levels With Coronary Artery Disease and Myocardial Infarction.” JAMA, vol. 318, no. 4, July 2017, pp. 371–80. PubMed, https://doi.org/10.1001/jama.2017.8981.
Li, Bowei, et al. “Infection and Atherosclerosis: TLR-Dependent Pathways.” Cellular and Molecular Life Sciences, vol. 77, no. 14, 2020, p. 2751. www.ncbi.nlm.nih.gov, https://doi.org/10.1007/s00018-020-03453-7.
Li, Fei, et al. “SARS-CoV-2 Spike Promotes Inflammation and Apoptosis through Autophagy by ROS-Suppressed PI3K/AKT/MTOR Signaling.” Biochimica Et Biophysica Acta. Molecular Basis of Disease, vol. 1867, no. 12, Dec. 2021, p. 166260. PubMed, https://doi.org/10.1016/j.bbadis.2021.166260.
Linton, MacRae F., et al. “The Role of Lipids and Lipoproteins in Atherosclerosis.” Endotext, edited by Kenneth R. Feingold et al., MDText.com, Inc., 2000. PubMed, http://www.ncbi.nlm.nih.gov/books/NBK343489/.
—. “The Role of Lipids and Lipoproteins in Atherosclerosis.” Endotext, edited by Kenneth R. Feingold et al., MDText.com, Inc., 2000. PubMed, http://www.ncbi.nlm.nih.gov/books/NBK343489/.
Liu, Jinxue, et al. “MiR-126-HMGB1-HIF-1 Axis Regulates Endothelial Cell Inflammation during Exposure to Hypoxia-Acidosis.” Disease Markers, vol. 2021, 2021. www.ncbi.nlm.nih.gov, https://doi.org/10.1155/2021/4933194.
—. “MiR-126-HMGB1-HIF-1 Axis Regulates Endothelial Cell Inflammation during Exposure to Hypoxia-Acidosis.” Disease Markers, vol. 2021, 2021. www.ncbi.nlm.nih.gov, https://doi.org/10.1155/2021/4933194.
Liu, Minxuan, et al. “Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1.” Frontiers in Physiology, vol. 11, 2020. Frontiers, https://www.frontiersin.org/article/10.3389/fphys.2020.00774.
—. “Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1.” Frontiers in Physiology, vol. 11, 2020. Frontiers, https://www.frontiersin.org/article/10.3389/fphys.2020.00774.
Malavige, Gathsaurie Neelika, and Graham S. Ogg. “Pathogenesis of Vascular Leak in Dengue Virus Infection.” Immunology, vol. 151, no. 3, July 2017, pp. 261–69. PubMed, https://doi.org/10.1111/imm.12748.
Moore, Kathryn J., et al. “Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part II).” Journal of the American College of Cardiology, vol. 72, no. 18, Oct. 2018, p. 2181. www.ncbi.nlm.nih.gov, https://doi.org/10.1016/j.jacc.2018.08.2147.
Rajendran, Peramaiyan, et al. “The Vascular Endothelium and Human Diseases.” International Journal of Biological Sciences, vol. 9, no. 10, 2013, p. 1057. www.ncbi.nlm.nih.gov, https://doi.org/10.7150/ijbs.7502.
Serhan, Charles N., et al. “Maresins: Novel Macrophage Mediators with Potent Antiinflammatory and Proresolving Actions.” The Journal of Experimental Medicine, vol. 206, no. 1, Jan. 2009, p. 15. www.ncbi.nlm.nih.gov, https://doi.org/10.1084/jem.20081880.
Silva, Grazielle Caroline, et al. “Experimental Periodontal Disease Triggers Coronary Endothelial Dysfunction in Middle-Aged Rats: Preventive Effect of a Prebiotic β-Glucan.” The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, vol. 76, no. 8, July 2021, pp. 1398–406. PubMed, https://doi.org/10.1093/gerona/glab066.
Topol, Eric J., et al. “Genetic Susceptibility to Myocardial Infarction and Coronary Artery Disease.” Human Molecular Genetics, vol. 15 Spec No 2, Oct. 2006, pp. R117-123. PubMed, https://doi.org/10.1093/hmg/ddl183.
Waltmann, Meaghan D., et al. “Apolipoprotein E Receptor-2 Deficiency Enhances Macrophage Susceptibility to Lipid Accumulation and Cell Death to Augment Atherosclerotic Plaque Progression and Necrosis.” Biochimica et Biophysica Acta, vol. 1842, no. 9, Sept. 2014, p. 1395. www.ncbi.nlm.nih.gov, https://doi.org/10.1016/j.bbadis.2014.05.009.
Xu, Lang-Biao, et al. “Rs10757274 Gene Polymorphisms in Coronary Artery Disease: A Systematic Review and a Meta-Analysis.” Medicine, vol. 99, no. 3, Jan. 2020, p. e18841. PubMed, https://doi.org/10.1097/MD.0000000000018841.
Yamamoto, Yasuhiro, et al. “Intermittent Local Periodontal Inflammation Causes Endothelial Dysfunction of the Systemic Artery via Increased Levels of Hydrogen Peroxide Concomitantly with Overexpression of Superoxide Dismutase.” International Journal of Cardiology, vol. 222, Nov. 2016, pp. 901–07. PubMed, https://doi.org/10.1016/j.ijcard.2016.08.099.
Zhang, Yefei, et al. “Resveratrol Prevents TNF-α-Induced VCAM-1 and ICAM-1 Upregulation in Endothelial Progenitor Cells via Reduction of NF-ΚB Activation.” The Journal of International Medical Research, vol. 48, no. 9, Sept. 2020. www.ncbi.nlm.nih.gov, https://doi.org/10.1177/0300060520945131.
Zhao, Zi-Wen, et al. “Circulating Soluble Lectin‐Like Oxidized Low‐Density Lipoprotein Receptor‐1 Levels Are Associated With Angiographic Coronary Lesion Complexity in Patients With Coronary Artery Disease.” Clinical Cardiology, vol. 34, no. 3, Mar. 2011, p. 172. www.ncbi.nlm.nih.gov, https://doi.org/10.1002/clc.20847.